Ang Ketonuria ay ang pagkakaroon ng ihi ng mga katawan ng ketone, lalo na ang acetone
Ang mekanismo ng ketonuria.
Sa isang malusog na tao, ang karbohidrat, lipid, at ilang mga protina ay na-oxidized sa carbon dioxide at tubig. Sa ilang mga kondisyon ng pathological, lalo na sa diyabetis, bumababa ang produksyon ng insulin. Sa atay, ang mga reserbang glycogen ay nabawasan, maraming mga tisyu ng katawan ang nasa isang estado ng gutom ng enerhiya. Sa ilalim ng mga kondisyong ito, ang mga proseso ng oksihenasyon ng mga protina at lipid sa atay ay isinaaktibo, ngunit ang kakulangan ng glycogen ay humantong sa kanilang hindi kumpletong oksihenasyon at akumulasyon sa dugo ng mga hindi natupok na mga produkto ng lipid at metabolismo ng protina - mga ketone na katawan. Dahil sa akumulasyon ng mga ketone na katawan sa dugo (ketonemia), ang pH ng dugo ay lumipat sa gilid ng acid. Ang kondisyong ito ay tinatawag na acidosis. Ang ihi ng naturang pasyente ay may isang matalim na reaksyon ng acid at amoy ng acetone.
Ang gutom, ang paggamit ng pangunahin na protina at mataba na pagkain, ang pagbubukod ng mga karbohidrat mula sa diyeta ay humantong sa pagtaas ng pagbuo ng mga ketone na katawan at ang kanilang pag-iipon sa ihi.
Sa isang maagang edad, ang ketonuria ay mas karaniwan kaysa sa mga matatanda at walang klinikal na kahalagahan. Ang kababalaghan na ito ay interesado sa pedyatrisyan na may "acetonemic pagsusuka" na nauugnay sa mga error sa diyeta.
Ang Ketonuria sa panahon ng postoperative ay dahil sa pagkasira ng protina dahil sa trauma ng kirurhiko.
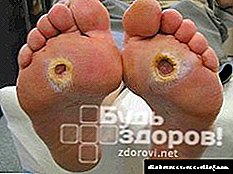
Sa mga may sapat na gulang, ang ketonuria ay nangyayari sa malubhang anyo ng diyabetis at may malaking halaga ng diagnostic. Sa mga bata, maaari itong makasama sa iba't ibang mga sakit, dahil sa kahusayan ng metabolismo ng karbohidrat. Samakatuwid, kahit na ang mga menor de edad na pagkakamali sa diyeta, lalo na sa pagkakaroon ng talamak na impeksiyon, pagkagambala sa nerbiyos, sobrang paggawa, atbp maaaring humantong sa ketosis. Ang Ketonuria sa maagang pagkabata ay maaaring sundin na may toxicosis, matagal na gastrointestinal na karamdaman, pagdidiyeta at iba pang mga sakit. Sa mga bagong silang, ang isang pagtaas ng mga ketones ng ihi ay halos palaging sanhi ng malnutrisyon. Ang Ketonuria, na sinusunod sa mga nakakahawang sakit - iskarlata na lagnat, trangkaso, tuberculous meningitis at pagkalasing, ay isang pangalawang palatandaan, lumilipas at walang mahusay na halaga ng diagnostic.
Ketonuria para sa diyabetis bubuo dahil sa tumaas na ketogenesis at kapansanan ketolysis. Ang pagtaas ng ketogenesis ay humantong sa pagtaas ng pagpapakilos ng mga taba mula sa adipose tissue, isang pagbawas sa pagbuo ng oxalacetate sa cycle ng Krebs, at isang pagbawas sa biosynthesis ng mga fatty acid. Sa malubhang diyabetis, na sinamahan ng pinsala sa tisyu ng bato (isang lugar para sa pagkasira ng mga ketones), nangyayari ang isang karagdagang paglabag sa ketolysis.
Ang kawalan ng glucosuria sa pagkakaroon ng ketonuria ay hindi kasama ang diabetes.
Karaniwan, ang mga katawan ng ketone ay nabuo sa isang maliit na halaga mula sa panghuling produkto ng karbohidrat at lipid metabolismo - acetyl-CoA (C2-bodies) sa pamamagitan ng acetoacetyl-CoA at halos ganap na neutralisado.
Sa diabetes mellitus, ang pagpapakilos ng mga lipid ay nagdaragdag sa pagbuo ng isang malaking halaga ng acetyl CoA (C2katawan). sa parehong oras, dahil sa isang paglabag sa metabolismo ng karbohidrat, isang pagbawas sa pagbuo ng oxalacetate, kung saan2Ang mga bodega ay kasama sa siklo ng Krebs at sumunog hanggang sa carbon dioxide at tubig. Bilang karagdagan, bilang isang resulta ng pagtaas ng lipid breakdown, ang reverse biosynthesis ng C ay naka-block.2katawan sa mga fatty acid. Sa gayon, ang isang malaking halaga ng C ay maipon2-Si sino, na humahantong sa paggawa ng isang malaking halaga ng CoA, at dahil dito acetone, acetoacetic at beta-hydroxybutyric acid, na kung saan ay excreted sa ihi.
Mga katawan ng ketone
Ito ay mga pansamantalang mga nabubulok na produkto na nabuo sa pag-andar ng atay. Karaniwan, ang metabolismo ay naglalantad ng mga katawan ng ketone upang higit na mapanghinawa.
Ang mga reserbang glucose sa katawan ay matatagpuan sa taba ng katawan, na ang dahilan kung bakit ang ketonuria ay isang malakas na kakulangan ng karbohidrat. Ang mga ketone na katawan ay isang mahusay na tagabigay ng enerhiya. Sa ito, kahit na ang mga fatty acid ay nahuli sa kanilang likuran. Samakatuwid, kapag ang utak o puso ay kulang sa mga acid ng phosphoric, ang katawan ay agad na gumagawa ng mga ketone na katawan sa isang pinabilis na bilis.
Ano ang mga dahilan
Ang Ketonuria ay ang nilalaman ng ihi ng mga ketone na katawan kaysa sa normal.
Sa isang malusog na katawan ng tao, ang metabolismo ay maayos na balanse. Ano ang maaaring maging sanhi ng ketonuria?

- Ang namamayani ng protina at taba sa diyeta. Dahil sa isang pagbaba ng mga karbohidrat sa pagkain, ang mga cell ay kulang sa nutrisyon. Laban sa background na ito, ang ketonuria ay bubuo. Ito ang reaksyon ng katawan sa isang kawalan ng timbang sa diyeta.
- Ang mga diyeta at pang-aabuso ng gutom ay maaaring mag-trigger ng hitsura ng mga ketone na katawan sa ihi. Ang mga pagkakamali sa pagpili ng mga diyeta ay humantong sa isang mabilis na pagsira ng mga taba, habang ang bilang ng mga enzyme ay nagdaragdag. Ang Ketonuria din ang hitsura ng acetone sa dugo. Karaniwan, kung ang isang tao ay gutom ng higit sa anim na araw, ang nilalaman ng mga ketone na katawan sa katawan ng tao ay nagdaragdag sa itaas ng normal.
- Pangpamanhid para sa operasyon.
- Diabetes mellitus. Sa kasong ito, ang ketonuria ay lilitaw paminsan-minsan. Ang mga katawan ng ketone lamang ay hindi ang sanhi ng sakit. Sa diabetes mellitus, inireseta ang isang karagdagang kurso ng karbohidrat at insulin.
- Pag-aalis ng tubig. Ito ay nangyayari kapag ang katawan ay overheats o isang komplikasyon ng diabetes.
- Ang sakit sa atay o malalang nakakahawang sakit (hal., Dysentery).
- Ang pag-unlad ng mga bukol sa digestive tract.
- Mga karamdaman sa pancreas.
- Pagkalason ng alkohol o sa pamamagitan ng naturang mga compound ng kemikal bilang posporus, tingga.
- Pinsala sa gitnang sistema ng nerbiyos, kaguluhan sa nerbiyos.
Tandaan na ang amoy ng acetone kapag huminga o umihi ay isang senyas upang makita ang isang doktor. Ito rin ay isang dahilan upang mabago ang pamumuhay at balanse sa nutrisyon.
Pansin sa mga bata
Ang klinika ay madalas na kinonsulta kung ang pagsusuka ng bata, ang amoy ng acetone. O kaya lumitaw ang gayong amoy sa ihi. At kahit na ito ay isang palatandaan ng ketonuria, hindi kinakailangan na isang sintomas ng isang malubhang sakit.

Kadalasan, ang lahat ay ipinaliwanag ng isang hindi perpektong sistema ng metabolic. Ang sanhi ng ketonuria sa mga bata ay isang kondisyon kung saan ginugol ang isang malaking halaga ng enerhiya. Karaniwan itong nangyayari kapag:
- emosyonal na kaguluhan
- nadagdagan ang pisikal na bigay,
- hindi balanseng diyeta
- isang malamig
Ang katotohanan ay ang katawan ng bata ay walang malalaking tindahan ng glycogen, samakatuwid, ang aktibong pagkasira ng mga taba ay nangyayari at ang mga palatandaan ng ketonuria ay sinusunod.
Pagbubuntis
Sa siyam na buwan ng pagbubuntis, ang mga kababaihan ay madalas na kumuha ng mga pagsusuri sa ihi. Ito ay kinakailangan upang hindi makaligtaan ang pinakamaliit na paglihis mula sa pamantayan sa katawan para sa buong panahon ng pagbubuntis. Sa katunayan, ang lahat ng mga organo ay may isang malaking pagkarga. Ano ang ibig sabihin na ang mga katawan ng ketone ay matatagpuan sa ihi?
Karaniwan ang kanilang maliit na nilalaman ay ang pamantayan. Ang pinakasimpleng pagsusuri ay pagpasa sa mabilis na pagsubok. Kailangan mong bawasan ang test strip sa ihi.
Ang isang negatibong pagsubok ay itinuturing na normal sa panahon ng pagbubuntis o ang bilang ng mga ketones ay minimal. Kung ang halaga ng pagsubok ay mula 15 hanggang 160 mg / dl - ito ang sanhi ng pag-aalala.
Sa panahon ng pagbubuntis, ang ketonuria ay isang nakababahala na sintomas. Lumilitaw ito sa maagang toxicosis. Ang pagkalason ng katawan, at samakatuwid ang fetus na may acetone, ay kumplikado ang kurso ng pagbubuntis.

Kung ang isang buntis ay may malubhang pagkabigo sa hormonal, nasa peligro siya.
Ang isang pagtaas sa antas ng ketones sa ihi ng isang bagong panganak ay dahil sa hindi sapat na pagpapakain o mga error sa nutrisyon.
Mga sintomas ng Ketonuria
Kung ang antas ng mga ketones sa katawan ay makabuluhang nadagdagan, pagkatapos ang ketonuria ay magpapakita mismo:
- nakakapagod na pag-aantok,
- amoy ng acetone mula sa bibig,
- ihi acetone amoy
- ang pagtatasa ay magpapakita ng isang mataas na antas ng mga puting selula ng dugo sa dugo,
- ang isang biochemical test ng dugo ay magpapakita ng isang mababang nilalaman ng glucose,
Kung hinikayat ng ketonuria ang isang tumalon sa dugo ng acetone, maaaring mangyari ang isang krisis sa acetone.
Ang isang makabuluhang pagtaas ng kaasiman sa mga cell ay maaaring malubhang makapinsala sa mga panloob na organo. Sa kasong ito, ang isang proteksyon na reaksyon ay inilunsad - pagsusuka.
Mga sintomas ng ketonuria sa mga bata:
- reklamo ng sakit sa tiyan
- mga reklamo ng sakit ng ulo
- ang bata ay pagod, nakakapagod,
- reklamo ng pagduduwal
- pagsusuka
- pagtaas ng temperatura sa 39 ° C,
- pagtanggi ng pagkain,
- amoy tulad ng acetone
- pinalaki ang atay
Maaari bang makita ang ketonuria?
Tanging ang pagtatasa ng kemikal ay maaaring makakita ng pagtaas sa antas ng mga ketone na katawan sa katawan. Tatalakayin agad ng laboratoryo ang pamantayan ng mga katawan ng ketone na nakapaloob dito.
Sa modernong gamot, ang ketonuria ay napansin:
- Lange test,
- Legal na pagsubok
- Sample Lestrade,
- binagong sample ng Rothera,
- mabilis na mga pagsubok

Ang mga mabilis na pagsubok, siyempre, ang pinaka-karaniwan. Ang kanilang pagkilos ay batay sa isang reaksyon ng kemikal, ang resulta kung saan makikita halos kaagad. Ang isa ay dapat lamang ilagay ang test strip sa ihi o i-drop ito sa isang test tablet. Sa kaso ng isang positibong reaksyon, ang mga pagsusuri ay lilang. Ang ningning ng kulay ay nagbibigay-daan sa iyo upang hatulan sa pamamagitan ng isang espesyal na sukat ng kulay tungkol sa kung magkano ang pamantayan ng mga katawan ng ketone ay lumampas.
International Classification ng mga Karamdaman
Ang International Statistical Classification of Diseases at Mga Kaugnay na Suliranin sa Kalusugan (ICD) ay isang gabay na sanggunian kung saan maihahambing ang mga materyales sa buong mundo. Sa tulong nito, ang iba't ibang mga pamamaraan ng pamamaraan ay pinagsama. Ang mga istatistika ng sakit ay naipon at inuri. Tuwing sampung taon, ang IBC ay susuriin ng isang komisyon ng World Health Organization. Upang pag-uri-uriin at pag-aralan ang buong masa ng naipon na mga resulta mula sa punto ng view ng mga istatistika, isinalin ito sa mga code ng alphanumeric. Sa ganoong gawain, ginagamit ang binuo na ICD. Ngayon tumutugma ito sa ICD-10. Naglalaman ito ng 22 mga klase (mga seksyon).
Ayon sa direktoryo ng ICD, ang ketonuria ay may code R82.4.
Pag-iwas at Pagkain ng Ketonuria
Para sa pag-iwas sa ketonuria, kinakailangan:
- kumain ng tama
- humantong sa isang malusog na pamumuhay:
- hangga't maaari upang maging sa sariwang hangin,
- katamtaman na ehersisyo
- Huwag simulan ang mga malalang sakit.

Sa isang talamak na sakit tulad ng diabetes, ang isang doktor ay kumonsulta sa sistematikong. Paminsan-minsan kinakailangan na kumuha ng isang ekspresyong pagsubok.
Ang isang positibong reaksyon ay nagpapahiwatig ng patolohiya sa katawan. Bigyang-pansin ang signal! Ang madaliang apela sa klinika ay mabilis na babalik sa normal ang katawan. Nang hindi umaalis sa diyeta na inireseta ng doktor, maaari mong mapabilis ang paggaling.
Mga prinsipyo sa pagkain ng Ketonuria:
- huwag kumain ng mga pagkaing mataas sa protina at taba,
- regular na paggamit ng karbohidrat
- uminom ng higit pang mga solusyon sa tubig at soda (na may diyabetis - insulin).
Ang menu ay dapat isama: pinakuluang karne ng baka at kuneho, iba't ibang mga sopas ng gulay, isda na mababa ang taba. Mga kapaki-pakinabang na butil na walang mantikilya, gulay at prutas. Inirerekomenda na uminom ng mas maraming mga juice, inumin ng prutas, compotes.
Ibukod mula sa menu:
- mataba na karne
- maanghang na mga panimpla
- matamis
- sitrus prutas
- saging
- kabute
- mabilis na pagkain.
Tumutok sa paggamot
Ang Ketonuria ay hindi dapat tratuhin bilang isang hiwalay na sakit. Siya lang ang kinahinatnan. Ito ay kinakailangan upang ibukod ang mga sanhi na sanhi nito. Una, kinakailangan ang isang kumpletong pagsusuri. Ang katumpakan ng diagnosis at ang pagtatatag ng sanhi ng ketonuria ginagarantiyahan ang matagumpay na paggamot.
Nagbibigay ang mga doktor ng ilang mga tip:
- Kung ikaw ay sobrang timbang, dapat mong ayusin ang mga araw ng pag-aayuno paminsan-minsan.
- Kung ang pagsusuri ay nagsiwalat ng pagtaas sa mga ihi keton, bumili ng mga pagsubok upang magamit ang mga ito sa bahay.

- Ang ihi ay dapat na nakolekta para sa pagsusuri nang hindi hihigit sa apat na oras bago ang paghahatid.
- Ang bata ay maaaring lasing na may inuming alkalina tuwing 10-15 minuto sa maliit na bahagi. Ang aktibong uling, enterosgel ay makakatulong upang limasin ang digestive tract.
- Sa paparating na pagsusuka, kapaki-pakinabang na uminom ng isang fractional na inumin. Tumawag ng isang ambulansya.
Paano gamutin ang ketonuria, may mga tip sa katutubong gamot.
- Sa madalas na pagsusuka, uminom ng mineral na tubig, pinatuyong compote ng prutas, solusyon sa glucose. Isang kutsara sa sampung minuto.
- Maglagay ng enema sa bahay. Una, ang tubig sa temperatura ng silid, kalaunan - mainit-init, kung saan idinagdag ang isang kutsarang soda.
- Kumuha ng inumin: matunaw ang 2 tbsp sa 1 litro ng tubig. honey, ibuhos ang juice ng isang lemon. Uminom ng 1 tbsp. tuwing 15 minuto.
- Recipe para sa solusyon ng soda: matunaw ang 1 kutsarita ng soda sa 250 ML ng tubig. Uminom ng 1 tsp. tuwing 10 minuto.
- Kumuha ng mga decoction ng nakapapawi na mga halamang gamot.
- Upang mapabilis ang pag-aalis ng mga lason mula sa katawan, kumain ng kaunti. Mas mainam na kumain lamang ng mga crackers.
Ang paggamot ay nakasalalay sa mga indibidwal na katangian ng katawan. Dapat itong mahigpit na kontrolado ng dumadating na manggagamot.
76. Kolesterol. Mga paraan ng pagpasok, paggamit at paglabas mula sa katawan. Serum kolesterol. Biosynthesis ng kolesterol, ang mga yugto nito. Ang regulasyon ng sintesis.
Ang Cholesterol ay isang steroid na tiyak sa mga organismo ng hayop. Ito ay synthesized sa maraming mga tisyu ng tao, ngunit ang pangunahing lugar ng synthesis ay ang atay. Sa atay, higit sa 50% ng kolesterol ay synthesized, sa maliit na bituka - 15-20%, ang natitirang kolesterol ay synthesized sa balat, adrenal cortex, at gonads. Halos 1 g ng kolesterol ay synthesized bawat araw sa katawan, 300-500 mg ang pinalamanan ng pagkain (Larawan 8-65). Ang Cholesterol ay gumaganap ng maraming mga pag-andar: ito ay isang bahagi ng lahat ng mga lamad ng cell at nakakaapekto sa kanilang mga pag-aari, nagsisilbing isang paunang substrate sa synthesis ng mga acid ng apdo at mga hormone ng steroid. Ang mga precursor sa metabolic pathway ng synthesis ng kolesterol ay lumiliko din sa ubiquinone, isang sangkap ng chain ng paghinga at dolichol, na kasangkot sa synthesis ng glycoproteins. Dahil sa grupong hydroxyl na ito, ang kolesterol ay maaaring mabuo ang mga ester na may mga fatty acid. Ang Etherified kolesterol ay namumuno sa dugo at nakaimbak sa maliit na dami sa ilang mga uri ng mga cell na ginagamit ito bilang isang substrate para sa synthesis ng iba pang mga sangkap. Ang kolesterol at ang mga ester nito ay mga molekulang hydrophobic, kaya dinadala sila ng dugo lamang bilang bahagi ng iba't ibang uri ng gamot. Ang pagpapalitan ng kolesterol ay sobrang kumplikado - para lamang sa synthesis nito, mga 100 magkakasunod na reaksyon ang kinakailangan. Sa kabuuan, halos 300 iba't ibang mga protina ang nasasangkot sa metabolismo ng kolesterol. Ang mga karamdaman ng metabolismo ng kolesterol ay humantong sa isa sa mga pinaka-karaniwang sakit - atherosclerosis. Ang dami ng namamatay mula sa mga epekto ng atherosclerosis (myocardial infarction, stroke) ay humantong sa pangkalahatang istraktura ng mortalidad. Ang Atherosclerosis ay isang "polygenic disease", i.e. maraming mga kadahilanan ay kasangkot sa pag-unlad nito, ang pinakamahalaga sa kung saan ay namamana. Ang akumulasyon ng kolesterol sa katawan ay humahantong sa pag-unlad ng isa pang karaniwang sakit - sakit sa bato.
A. Sintesis ng kolesterol at regulasyon nito
Ang mga reaksyon ng synthesis ng kolesterol ay nangyayari sa cytosol ng mga selula. Ito ang isa sa pinakamahabang metabolic pathway sa katawan ng tao.
Phenylketonuria

Phenylketonuria - isang namamana na paglabag sa metabolismo ng amino acid dahil sa kakulangan ng mga enzyme ng atay na kasangkot sa metabolismo ng phenylalanine sa tyrosine. Ang mga maagang palatandaan ng phenylketonuria ay pagsusuka, nakamamatay o hyperactivity, ang amoy ng amag mula sa ihi at balat, naantala ang pag-unlad ng psychomotor, mga tipikal na huli na mga palatandaan ay kasama ang oligophrenia, pagkaantala sa pagpapaunlad ng pisikal, pagkukumbinsi, mga pagbabago sa balat ng eczematous, atbp. kasunod na mga diagnostic ay nagsasama ng molekula na pagsusuri ng molekula, pagpapasiya ng konsentrasyon ng phenylalanine ng dugo, pagsusuri ng biochemical ng ihi, EEG, at MRI ng utak. Ang paggamot ng phenylketonuria ay upang sundin ang isang espesyal na diyeta.

Pangkalahatang impormasyon
Ang Phenylketonuria (sakit ng Felling, phenylpyruvic oligophrenia) ay isang congenital, genetically na tinutukoy na patolohiya na nailalarawan sa pamamagitan ng kapansanan na hydroxylation ng phenylalanine, ang akumulasyon ng mga amino acid at metabolites sa mga physiological fluids at tisyu, na sinusundan ng matinding pinsala sa sentral na sistema ng nerbiyos. Ang Phenylketonuria ay unang inilarawan ni A. Felling noong 1934, nangyayari ito sa dalas ng 1 kaso bawat 10,000 mga bagong panganak. Sa panahon ng neonatal, ang phenylketonuria ay walang mga klinikal na pagpapakita, gayunpaman, ang paggamit ng phenylalanine na may pagkain ay nagiging sanhi ng paghahayag ng sakit sa unang kalahati ng buhay, at pagkatapos ay humahantong sa malubhang sakit sa pag-unlad ng bata. Iyon ang dahilan kung bakit ang pre-sintomas na pagtuklas ng phenylketonuria sa mga bagong silang ay ang pinakamahalagang gawain ng neonatology, pediatrics at genetics.
Mga Sanhi ng Phenylketonuria
Ang Phenylketonuria ay isang autosomal na uring sakit na mana. Nangangahulugan ito na para sa pagbuo ng mga klinikal na palatandaan ng phenylketonuria, ang bata ay dapat magmana ng isang may sira na kopya ng gene mula sa parehong mga magulang, na mga heterozygous carriers ng mutant gene.
Kadalasan, ang pagbuo ng phenylketonuria ay sanhi ng isang mutation sa gene na naka-encode ng phenylalanine-4-hydroxylase enzyme na matatagpuan sa mahabang braso ng chromosome 12 (locus 12q22-q24.1). Ito ang tinatawag na klasikal na uri na phenylketonuria, na nagkakahalaga ng 98% ng lahat ng mga kaso ng sakit. Ang Hyperphenylalaninemia ay maaaring umabot ng 30 mg% at mas mataas. Kung hindi mababago, ang variant ng phenylketonuria na ito ay sinamahan ng malalim na pag-retard sa kaisipan.
Bilang karagdagan sa klasikal na form, ang atypical phenylketonuria ay nakikilala, na nagpapatuloy sa parehong mga klinikal na sintomas, ngunit hindi matapat sa pagwawasto sa pamamagitan ng therapy sa diyeta. Kabilang dito ang uri ng phenylketonuria II (dehydroterterin kakulangan ng reductase), uri ng phenylketonuria III (kakulangan tetrahydrobiopterin) at iba pa, mas bihirang mga variant.
Ang posibilidad na manganak ng isang bata na may phenylketonuria ay nagdaragdag na may malapit na pag-aasawa.
Pathogenesis ng phenylketonuria
Ang klasikal na anyo ng phenylketonuria ay batay sa kakulangan ng phenylalanine-4-hydroxylase enzyme na kasangkot sa pag-convert ng phenylalanine sa tyrosine sa hepatocyte mitochondria. Kaugnay nito, ang tyrosine derivative tyramine ay ang panimulang materyal para sa synthesis ng catecholamines (adrenaline at norepinephrine), at diiodotyrosine para sa pagbuo ng thyroxine. Bilang karagdagan, ang pagbuo ng melanin pigment ay ang resulta ng metabolismo ng phenylalanine.
Ang kakulangan sa heneral ng phenylalayin-4-hydroxylase enzyme sa phenylketonuria ay humahantong sa isang paglabag sa oksihenasyon ng phenylalanine mula sa pagkain, na nagreresulta sa konsentrasyon nito sa dugo (phenylalaninemia) at cerebrospinal fluid na nagdaragdag nang malaki, at ang antas ng tyrosine ay bumababa nang naaayon. Ang labis na phenylalanine ay tinanggal sa pamamagitan ng pagtaas ng pag-ihi ng ihi ng mga metabolites - phenylpyruvic acid, phenylmilactic at phenylacetic acid.
Ang pagkagambala ng metabolismo ng amino acid ay sinamahan ng kapansanan na myelination ng mga fibers ng nerve, isang pagbawas sa pagbuo ng mga neurotransmitters (dopamine, serotonin, atbp.), Na nag-trigger ng mga mekanismo ng pathogenetic ng pag-iisip ng retardation at progresibong demensya.
Sintomas ng phenylketonuria
Ang mga bagong panganak na may phenylketonuria ay walang mga klinikal na palatandaan ng sakit. Karaniwan, ang paghahayag ng phenylketonuria sa mga bata ay nangyayari sa edad na 2-6 na buwan. Sa simula ng pagpapakain, ang protina ng gatas ng suso o ang mga kapalit nito ay nagsisimula na pumasok sa katawan ng sanggol, na humahantong sa pagbuo ng una, hindi tiyak na mga sintomas - nakamamatay, minsan pagkabalisa at hyper excitability, regurgitation, muscular dystonia, convulsive syndrome. Ang isa sa mga unang palatandaan ng pathognomonic ng phenylketonuria ay patuloy na pagsusuka, na kung saan ay madalas na nagkakamali na itinuturing na isang paghahayag ng pyloric stenosis.
Sa ikalawang kalahati ng taon, ang lagas ng bata sa pag-unlad ng psychomotor ay napansin. Ang bata ay nagiging hindi gaanong aktibo, walang malasakit, tumitigil na makilala ang mga mahal sa buhay, hindi subukang umupo at tumayo sa kanyang mga paa. Ang hindi normal na komposisyon ng ihi at pawis ay nagdudulot ng isang katangian na "mouse" na amoy (ang amoy ng amag) na nagmumula sa katawan. Kadalasan mayroong pagbabalat ng balat, dermatitis, eksema, scleroderma.
Sa mga bata na may phenylketonuria na hindi tumatanggap ng paggamot, microcephaly, prognathia, sa paglaon (pagkatapos ng 1.5 taon) teething, enamel hypoplasia ay napansin. Ang pagkaantala sa pagbuo ng pagsasalita ay nabanggit, at sa pamamagitan ng 3-4 na taon na malalim na oligophrenia (idiocy) at isang halos kumpletong kawalan ng pagsasalita ay napansin.
Ang mga bata na may phenylketonuria ay may isang dysplastic na pangangatawan, madalas na mga depekto sa congenital heart, autonomic dysfunctions (pagpapawis, acrocyanosis, arterial hypotension), at nagdurusa mula sa pagkadumi. Ang mga katangian ng phenotypic ng mga batang nagdurusa sa phenylketonuria ay may kasamang magaan na balat, mata at buhok. Ang isang bata na may phenylketonuria ay nailalarawan ng isang tiyak na magpose ng isang "sastre" (itaas at mas mababang mga limbong baluktot sa mga kasukasuan), panginginig ng kamay, isang wobbly, mincing gait, at hyperkinesis.
Ang mga klinikal na pagpapakita ng uri II phenylketonuria ay nailalarawan sa pamamagitan ng isang matinding antas ng pag-retard sa pag-iisip, nadagdagan ang pagkamayamutin, mga seizure, spastic tetraparesis, at tendon hyperreflexia. Ang pag-unlad ng sakit ay maaaring humantong sa pagkamatay ng isang bata na may edad 2 hanggang 3 taon.
Kapag ang uri ng phenylketonuri III ay bubuo ng isang triad ng mga palatandaan: microcephaly, oligophrenia, spastic tetraparesis.
Diagnosis ng phenylketonuria
Sa kasalukuyan, ang diagnosis ng phenylketonuria (pati na rin ang galactosemia, congenital hypothyroidism, adrenogenital syndrome at cystic fibrosis) ay bahagi ng programa ng neonatal screening para sa lahat ng mga bagong silang.
Ang isang screening test ay isinasagawa sa 3-5 araw ng buhay ng isang full-term at 7 araw ng buhay ng isang napaaga na sanggol sa pamamagitan ng pagkuha ng isang sample ng dugo ng capillary sa isang espesyal na form ng papel. Kung ang hyperphenylalanemia ay napansin, higit sa 2.2 mg% ng bata ang tinukoy sa mga genetika ng pediatric para sa muling pagsusuri.
Upang kumpirmahin ang diagnosis ng phenylketonuria, ang konsentrasyon ng phenylalanine at tyrosine sa dugo ay nasuri, ang aktibidad ng hepatic enzymes (phenylalanine hydroxylase) ay natutukoy, isang pagsusuri ng biochemical ng ihi (pagpapasiya ng mga ketonic acid), ang mga metabolites ng catecholamines sa ihi, atbp ay isinasagawa.
Ang isang genetic na depekto sa phenylketonuria ay maaaring napansin kahit sa pagbubuntis sa panahon ng isang nagsasalakay prenatal diagnosis ng pangsanggol (chorionbiopsy, amniocentesis, cordocentesis).
Ang pagkakaiba-iba ng diagnosis ng phenylketonuria ay isinasagawa kasama ang intracranial birth trauma sa mga bagong panganak, impeksyon sa intrauterine, at iba pang mga metabolikong karamdaman ng mga amino acid.
Paggamot Phenylketonuria
Ang isang pangunahing kadahilanan sa paggamot ng phenylketonuria ay isang diyeta na pinipigilan ang paggamit ng protina sa katawan. Inirerekomenda ang paggamot upang magsimula sa isang konsentrasyon ng phenylalanine> 6 mg%. Ang mga espesyal na mixtures ay binuo para sa mga sanggol - Afenilak, Lofenilak, para sa mga bata na higit sa 1 taong gulang - Tetrafen, Phenyl-free, higit sa 8 taong gulang - Maxamum-XP at iba pa.Ang batayan ng diyeta ay mga pagkaing mababa ang protina - prutas, gulay, juice, protina hydrolysates at amino acid mixtures . Ang pagpapalawak ng diyeta ay posible pagkatapos ng 18 taon na may kaugnayan sa isang pagtaas sa pagpapaubaya sa phenylalanine. Alinsunod sa batas ng Russia, ang pagbibigay ng nutrisyon sa medikal para sa mga taong nagdurusa sa phenylketonuria ay dapat na walang bayad.
Ang mga pasyente ay inireseta ng paggamit ng mga compound ng mineral, bitamina ng grupo B, atbp, ayon sa mga indikasyon - mga gamot na nootropic, anticonvulsants. Sa kumplikadong therapy ng phenylketonuria, pangkalahatang masahe, ehersisyo therapy, at acupuncture ay malawakang ginagamit.
Ang mga bata na nagdurusa sa phenylketonuria ay nasa ilalim ng pangangasiwa ng isang lokal na pedyatrisyan at neuropsychiatrist, at madalas na nangangailangan ng tulong ng isang speech therapist at isang pathologist. Maingat na pagsubaybay sa katayuan ng neuropsychic ng mga bata, ang pagsubaybay sa antas ng phenylalanine sa dugo at mga tagapagpahiwatig ng electroencephalogram.
Ang mga diypical form ng phenylketonuria na hindi matapat sa paggamot sa diyeta ay nangangailangan ng appointment ng mga hepatoprotectors, anticonvulsants, kapalit na therapy na may levodopa, 5-hydroxytryptophan.
Prediksyon at pag-iwas sa phenylketonuria
Ang pagsasagawa ng isang mass screening para sa phenylketonuria sa panahon ng neonatal ay nagbibigay-daan sa iyo upang ayusin ang maagang diet therapy at maiwasan ang malubhang pinsala sa cerebral, may kapansanan sa pag-andar ng atay. Sa maagang pag-appointment ng isang pag-aalis ng diyeta sa klasikal na phenylketonuria, mabuti ang pagbabala sa pag-unlad ng bata. Sa pamamagitan ng huli na paggamot, ang pagbabala para sa pag-unlad ng kaisipan ay mahirap.
Ang pag-iwas sa mga komplikasyon ng phenylketonuria ay binubuo sa pag-screening ng mga bagong panganak, maagang inireseta at pangmatagalang pagsunod sa nutrisyon sa pagdidiyeta.
Upang masuri ang panganib ng pagsilang sa isang bata na may phenylketonuria, ang paunang genetic counseling ay dapat ibigay sa mga mag-asawa na mayroon nang isang may sakit na bata, ay nasa isang magkakasamang relasyon, at may mga kamag-anak na may sakit na ito. Ang mga kababaihan na may phenylketonuria na nagpaplano ng pagbubuntis ay dapat sundin ang isang mahigpit na diyeta bago ang paglilihi at sa panahon ng pagbubuntis upang ibukod ang isang pagtaas sa antas ng phenylalanine at mga metabolites at may kapansanan sa pagbuo ng isang genetically malusog na fetus. Ang panganib ng pagkakaroon ng isang bata na may phenylketonuria sa mga magulang na nagdadala ng isang may sira na gene ay 1: 4.





-
Diosmin: mga tagubilin, komposisyon, presyo, mga pagsusuri
Ang mga tagubilin sa Diosmin para sa paggamit, presyo, mga pagsusuri, mga analogue ng gamot Paano kumpleto at permanenteng mapupuksa ang mga varicose veins! Ang mga varicose veins ay nagdudulot ng malubhang komplikasyon at mga kahihinatnan. Mayroong isang paraan na makakatulong na mapupuksa ang mga varicose veins magpakailanman. ... -

-
-